2026
40. A high surface area silica-supported aldehyde as a flow-compatible amine scavenger
A. MIchelle Reinhardt, Jenny-Lee Panayides and Darren Lyall Riley, RSC Advances, 2026, 16, 33, 30916-309227
I.F. 6.100
Article Citations

A silica-supported aldehyde scavenger () was prepared from silica by sequential NaOH activation, alkylation with 1,2-dichloroethane, and Kornblum oxidation of the resulting alkyl chloride in DMSO/K2CO3. The material was characterised by FTIR, PXRD, TGA/DSC, SEM, and BET physisorption, confirming successful surface functionalisation and a dramatic increase in surface area upon imine formation with probe amines. Loading capacity was experimentally determined to be 0.43–1.31 mmol g−1, comparable to commercial polystyrene-aldehyde resins. A D-optimal design of experiments (22 runs) evaluated the effects of solvent type (mapped by PCA), flow rate (0.1–1.0 mL min−1), temperature (30–90 °C), and amine concentration (0.2–1.0 M) on scavenging efficiency. Only solvent type had a significant influence on performance: hydrophobic solvents afforded near-quantitative scavenging, while polar non-polarisable solvents (notably acetonitrile) reduced efficiency to approximately 75% due to a solvation screening mechanism. The scavenger is flow-compatible and demonstrates utility as an in-line purification tool for continuous flow synthesis of small-molecule libraries.
39. Advances in solid handling for continuous flow synthesis of specialty chemicals and pharmaceuticals
Zen Johnston, Thabo Peme, Tommy Mabasa, Christopher Len, Darren Riley, Jenny-Lee Panayides, Cloudius Sagandira, Communications Chemistry, 2026, 9, 101
I.F. 5.900
Article Citations (1)

Continuous flow chemistry has transformed the synthesis of pharmaceuticals and specialty chemicals by advancing sustainability, efficiency, and process control. Despite these advantages, the management of solids remains a major challenge, often leading to clogging, inefficient mixing, and limitations in scalability. This review discusses recent strategies developed to overcome these obstacles, including the use of continuous stirred-tank reactors, packed-bed reactors with immobilized reagents, reaction design modifications, Pickering emulsions, colloidal nanoparticle suspensions, and specialised equipment such as agitated tubular reactors, spinning disk reactors, and sonicated systems. By critically assessing these developments, we chart the trajectory toward more resilient and robust flow-based manufacturing, consolidating continuous flow chemistry as a cornerstone of modern chemical manufacturing.
38. Revitalised Hofmann carbylamine synthesis made possible with flow chemistry
Zen Johnston, Jaimee Jugmohan, Jenny-Lee Panayides and Darren Lyall Riley, Reaction Chemistry and Engineering, 2026, 11, 42-48
I.F. 2.900


Isocyanides are of relevance to several scientific fields; however, over the last 150 years only a limited number of synthetic strategies have been reported for preparing them. In a newly developed flow approach, a neglected method for preparing isocyanides, the Hofmann carbylamine reaction, has been revisited and revitalised. The approach developed afforded the preparation of a diverse library of isocyanides in good conversions while only requiring a 15 min residence time at 70 °C. In addition, the method is operationally easy to apply, and it affords several advantages over the more commonly employed strategy of preparing isocyanides which involves the conversion of amines to formamides followed by dehydration to an isocyanide.
37. Natural acids as catalysts for the continuous flow production of the green solvent 2,2,5,5-tetramethyltetrahydrofuran
Bernice Mercia Currie, Estefan van Vuuren, Jaimee Jugmohan, Jenny-Lee Panayides and Darren Lyall Riley, Tetrahedron Green Chemistry, 2026, 7, 100089
I.F. 2.550

As the demand for chemists to adhere to green chemistry principles increases, so does the demand for green solvents. Unfortunately, many green solvents, such as 2,2,5,5-tetramethyltetrahydrofuran (TMTHF), are costly and difficult to source. Traditional synthesis of TMTHF from 2,5-dimethyl-2,5-hexanediol has been reported to be catalysed by acids such as phosphoric and sulfuric acid, or, more recently, by H-beta zeolite. Although H-beta zeolite catalysts are high-yielding and selective, the energy required for their regeneration is high, and their production has questionable environmental impacts. A new approach was developed using flow technologies and naturally occurring acids as catalysts for TMTHF synthesis. Flow technologies are scalable, safe, efficient, and reproducible for daily chemical reactions, aligning with principles of green chemistry. This study observed several key improvements, including i) the use of a natural acid as a catalyst, ii) the use of water as a solvent, and iii) a continuous process for multigram-scale synthesis of TMTHF using citric acid monohydrate, with a yield of 72%, resulting in a throughput of 8.24 g h-1 (9.43 kg L -1 h-1 space-time yield).
2025
36. Synthesis of an 8-membered oxygen-containing benzo-fused heterocycle using flow technologies – an exercise in undertaking research with sustainability as a driver
Bernice Mercia Currie, Nicole Candice Neyt, Tanya Olivier, Petra van der Merwe, Lerato Shirley Dibokwane, Ansche Michelle Reinhardt, Lorinda Tarien van Wyk, Jenny-Lee Panayides and Darren Lyall Riley, RSC Sustainability, 2025, 3, 1356-1365
I.F. 4.900


Due to their natural abundance and biological properties, benzo-fused heterocycles are attractive targets in the field of drug discovery. Previously, a synthetic strategy for accessing 5-, 6-, 7- and 8-membered oxygen-containing benzo-fused heterocycles with the oxygen atom in the less commonly encountered 2-position was reported, however, the approach was hindered by long reaction times and a reliance on high boiling point solvents such as DMF. Targeting an 8-membered analogue as an exemplar, we highlighted that the adoption of basic green chemistry principles coupled with the use of flow chemistry techniques could be utilised (with limited development time) to improve day-to-day sustainability when performing synthetic research. In the case in hand, several key improvements were noted including i) a higher overall yield (37 % vs. 26 %), ii) a significantly reduced reaction time (110 min vs. 136 h) and iii) the avoidance of the undesirable solvent DMF.
2024
35. The role of silicon in drug discovery: a review
Jenny-Lee Panayides, Darren Lyall Riley, Felix Hasenmaile and Willem A.L. van Otterlo, RSC Medicinal Chemisty, 2024, 15, 3286-3344
I.F. 4.200


This review aims to highlight the role of silicon in drug discovery. Silicon and carbon are often regarded as being similar with silicon located directly beneath carbon in the same group in the periodic table. That being noted, in many instances a clear dichotomy also exists between silicon and carbon, and these differences often lead to vastly different physiochemical and biological properties. As a result, the utility of silicon in drug discovery has attracted significant attention and has grown rapidly over the past decade. This review showcases some recent advances in synthetic organosilicon chemistry and examples of the ways in which silicon has been employed in the drug-discovery field.
34. The Synthesis of Bupropion Hydrochloride Under Greener and Safer Conditions Utilizing Flow Technologies
Lorinda T. Van Wyk, Nicole C. Neyt, Jaimee Jugmohan, Jenny-Lee Panayides and Darren L. Riley, Reaction Chemistry and Engineering, 2024, 9, 45-57
I.F. 3.400


Globally, major depressive disorders are a leading cause of disconsolateness affecting more than 300 million individuals. Bupropion is a unique dopamine-norepinephrine reuptake inhibitor (DNRI) commonly utilized in the treatment of depression, smoking cessation, ADHD and other addictions. Herein, we report our attempts to develop a greener, safer and more sustainable process for the preparation of bupropion hydrochloride employing flow chemistry. The use of obnoxious and corrosive liquid bromine was evaded through the employment of polymer-bound pyridinium tribromide and environmentally questionable solvents NMP and DMF were substituted with greener co-solvent systems with appreciable success. The final telescoped flow process afforded bupropion hydrochloride in a 69% overall yield, with improved process mass intensity, productivity and purity, as well as a reduction in reagents/solvents designated as red or amber in terms of H-codes.
2023
33. Large-Scale Flow Chemistry in Mechanochemistry and Emerging Technologies for Sustainable Chemical Manufacturing
Nicole Neyt, Jaimee Jugmohan, Wessel Bonnet, Jenny-Lee Panayides and Darren L. Riley, 2023, CRC Press
Book Chapter Citations

The chapter introduces the basics behind flow chemistry highlighting the relative advantages that the technology provides over batch-based chemistry. Upscaling of flow reactors is underpinned by an understanding of fluid dynamics and how it influences mixing time, residence time distribution, pressure drop, mass transfer and heat transfer. Under flow conditions scaling-up of tubular microreactors is achieved through the use of numbering-up, scaling-out and sizing-up strategies and thereafter downstream processing can be achieved using a number of approaches with the most robust being extractions using membrane technologies. Upscaled examples are described by highlighting the use of flow chemistry for cGMP manufacturing, photochemistry, electrochemistry, multiphasic chemistry and performing hazardous chemistries.
2022
32. The in silico and in vitro analysis of donepezil derivatives for Anopheles acetylcholinesterase inhibition
Thankhoe A. Rants’o , Divan G. van Greunen, C. Johan van der Westhuizen, Darren L. Riley, Jenny-Lee Panayides, Lizette L. Koekemoer, Robyn L. van Zyl. PLoS One, 2022, 17(11):e0277363
I.F. 3.240

Current studies on Anopheles anticholinesterase insecticides are focusing on identifying agents with high selectivity towards Anopheles over mammalian targets. Acetylcholinesterase (AChE) from electric eel is often used as the bioequivalent enzyme to study ligands designed for activity and inhibition in human. In this study, previously identified derivatives of a potent AChE, donepezil, that have exhibited low activity on electric eel AChE were assessed for potential AChE-based larvicidal effects on four African malaria vectors; An. funestus, An. arabiensis, An. gambiae and An. coluzzii. This led to the identification of four larvicidal agents with a lead molecule, 1-benzyl-N-(thiazol-2-yl) piperidine-4-carboxamide 2 showing selectivity for An. arabiensis as a larvicidal AChE agent. Differential activities of this molecule on An. arabiensis and electric eel AChE targets were studied through molecular modelling. Homology modelling was used to generate a three-dimensional structure of the An. arabiensis AChE for this binding assay. The conformation of this molecule and corresponding interactions with the AChE catalytic site was markedly different between the two targets. Assessment of the differences between the AChE binding sites from electric eel, human and Anopheles revealed that the electric eel and human AChE proteins were very similar. In contrast, Anopheles AChE had a smaller cysteine residue in place of bulky phenylalanine group at the entrance to the catalytic site, and a smaller aspartic acid residue at the base of the active site gorge, in place of the bulky tyrosine residues. Results from this study suggest that this difference affects the ligand orientation and corresponding interactions at the catalytic site. The lead molecule 2 also formed more favourable interactions with An. arabiensis AChE model than other Anopheles AChE targets, possibly explaining the observed selectivity among other assessed Anopheles species. This study suggests that 1-benzyl-N-(thiazol-2-yl) piperidine-4-carboxamide 2 may be a lead compound for designing novel insecticides against Anopheles vectors with reduced toxic potential on humans.
31. Assessing a sustainable manufacturing route to lapatinib
Roderick T. Stark, Domnic R. Pye, Wenyi Chen, Oliver J. Newton, Benjamin J. Deadman, Philip W. Miller, Jenny-Lee Panayides, Darren L. Riley, Klaus Hellgardt and King Kuok (Mimi) Hii, Reaction Chemistry and Engineering, 2022, 7, 2420-2426
I.F. 5.200

A synthetic route to an anti-cancer drug, lapatinib, was devised to support the development of a sustainable manufacturing process in South Africa. Quantitative metrics were employed to evaluate the sustainability of the key steps of the reaction.
30. Use of open-source software platform to develop dashboards for control and automation of flow chemistry equipment
C. Johan van der Westhuizen, Jurie Du Toit, Nicole. C. Neyt, Darren L. Riley and Jenny-Lee Panayides, Digital Discovery, 2022, 1, 596-604
I.F. 6.200


We report the development of an open-source software approach to monitor and control flow chemistry reactors from any smart device. The dashboard server can be run on a low-cost Raspberry Pi. Parameters of the equipment can be saved to a database for review in real-time or at a later stage. The dashboard control platform was demonstrated by performing the allylation of isovanillin in an autonomous closed-loop optimisation fashion using Summit for the optimisation algorithm. The space time yield (STY) was optimised for this reaction using a single-objective Bayesian optimisation (SOBO) approach. The best STY result of 791 g.dm-3.h-1 was achieved with the experimental conditions of 77.3 °C for the reactor column, a residence time of 4 min, with 1.005 equivalence of allyl bromide. All the software utilised in this paper was open-source, a tutorial-based approach to developing the program is included in the paper demonstrating the software capabilities.
29. Design and testing of an ozonolysis reactor module with on-the-fly ozone degassing under flow conditions
Nicole.C. Neyt, C. Johan van der Westhuizen, Jenny-Lee Panayides and Darren L. Riley, Reaction Chemistry and Engineering, 2022, 7, 1718-1727
I.F. 5.200


Ozonolysis is an attractive, efficient, and green means of introducing oxygen containing functionalities using only oxygen and electricity. Unfortunately, safety issues associated with the accumulation of dissolved ozone and potentially explosive ozonides coupled with an oxygen rich reaction atmosphere have limited its integration into large scale process reactions. Herein we report on the development and testing of a prototype flow-based ozonolysis reactor which allows on-the-fly removal of ozone and oxygen negating the need for a downstream degassing step and allowing the continuous processing of intermediate ozonides in a safe manner. The approach lends itself to being able to telescope directly into downstream reactions without concern for the effect of residual ozone and minimises contact between the oxygen rich ozone atmosphere and the reaction mixture. The prototype was shown to remove between 98.5 and 99.7% of residual ozone-oxygen on-the-fly and its performance was demonstrated through the ozonolysis of several alkenes to afford a range of oxygen containing functional groups in good to high yields.
28. Discovery of Novel Acetylcholinesterase Inhibitors by Virtual Screening, In Vitro Screening, and Molecular Dynamics Simulations
C. Johan van der Westhuizen, Andre’ Stander, Jenny-Lee Panayides and Darren L. Riley, Journal of Chemical Information and Modelling, 2022, 62(6) 1550-1572
I.F. 6.162


Alzheimer’s disease is the most common neurodegenerative disease and currently poses a significant socioeconomic problem. This study describes the uses of computer-aided drug discovery techniques to identify novel inhibitors of acetylcholinesterase, a target for Alzheimer’s disease. High-throughput virtual screening was employed to predict potential inhibitors of acetylcholinesterase. Validation of enrichment was performed with the DUD-E data set, showing that an ensemble of binding pocket conformations is critical when a diverse set of ligands are being screened. A total of 720 compounds were submitted for in vitro screening, which led to 25 hits being identified with IC50 values of less than 50 μM. The majority of these hits belonged to two scaffolds: 1-ethyl-3-methoxy-3-methylpyrrolidine and 1H-pyrrolo[3,2-c]pyridin-6-amine both of which are noted to be promising compounds for further optimization. As various possible binding poses were suggested from molecular docking, molecular dynamics simulations were employed to validate the poses. In the case of the most active compounds identified, a critical, stable water bridge formed deep within the binding pocket was identified potentially explaining in part the lack of activity for subsets of compounds that are not able to form this water bridge.
27. A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction
Ignacy Cukrowski, George Dhimba and Darren L. Riley, Molecules, 2022, 27(3), 962
I.F. 4.927

Modelling of the proline (1) catalyzed aldol reaction (with acetone 2) in the presence of an explicit molecule of dimethyl sulfoxide (DMSO) (3) has showed that 3 is a major player in the aldol reaction as it plays a double role. Through strong interactions with 1 and acetone 2, it leads to a significant increase of energy barriers at transition states (TS) for the lowest energy conformer 1a of proline. Just the opposite holds for the higher energy conformer 1b. Both the ‘inhibitor’ and ‘catalyst’ mode of activity of DMSO eliminates 1a as a catalyst at the very beginning of the process and promotes the chemical reactivity, hence catalytic ability of 1b. Modelling using a Molecular-Wide and Electron Density-based concept of Chemical Bonding (MOWED-CB) and the Reaction Energy Profile–Fragment Attributed Molecular System Energy Change (REP-FAMSEC) protocol has shown that, due to strong intermolecular interactions, the HN-C-COOH (of 1), CO (of 2), and SO (of 3) fragments drive a chemical change throughout the catalytic reaction. We strongly advocate exploring the pre-organization of molecules from initially formed complexes, through local minima to the best structures suited for a catalytic process. In this regard, a unique combination of MOWED-CB with REP-FAMSEC provides an invaluable insight on the potential success of a catalytic process, or reaction mechanism in general. The protocol reported herein is suitable for explaining classical reaction energy profiles computed for many synthetic processes.
* Special Issue – Advances in Theoretical and Computational Chemistry
2021
26. Rapid formation of 2-lithio-1-(triphenylmethyl)imidazole and substitution reactions in flow
Simeng Wang, Jenny-Lee Panayides, Darren Riley, Christopher J. Tighe, Klaus Hellgardt, King Kuok (Mimi) Hii and Philip W. Miller, Reaction Chemistry and Engineering, 2021, 6, 2018-2023
I.F. 4.239

The functionalisation of imidazoles is a necessary step in the formation of many active pharmaceutical intermediates. Herein, we report a flow chemistry approach for the rapid and efficient formation of 2-lithio-1-(triphenylmethyl)imidazole at ambient temperature and its reaction with a range of electrophiles, achieving modest to high yields (40–94%) in short reaction times (<1 min). The method is amenable to the scale-up of this highly reactive lithio-imidazole intermediate.
25. Application of reactor engineering concepts in continuous flow chemistry: a review
Nicole C. Neyt and Darren L. Riley, Reaction Chemistry and Engineering, 2021, 6, 1295-1326
I.F. 4.239


The adoption of flow technology for the manufacture of chemical entities, and in particular pharmaceuticals, has seen rapid growth over the past two decades with the technology now blurring the lines between chemistry and chemical engineering. Current indications point to a future in which flow chemistry and related technologies will be a major player in modern chemical manufacturing and the 4th industrial revolution. In this review we highlight the application of new reactor configurations and designs in the context of either bespoke or commercial flow apparatus specifically related to microwave chemistry, photochemical transformations, electrochemical promoted reactions and multi-phasic reactions. In addition, we look at how 3D printing in reactor design and computer-aided automation is growing within the field and finally describe how innovative solutions are being developed to tackle challenging down-stream processing operations.
24. Improved batch and flow syntheses of the nonsteroidal anti-inflammatory COX-2 inhibitor celecoxib
Chantal Scholtz and Darren L. Riley, Reaction Chemistry and Engineering, 2021, 6, 138-146
I.F. 4.239

The comparison of an improved conventional batch mode synthesis of the nonsteroidal anti-inflammatory COX-2 inhibitor celecoxib with its flow chemistry alternative is reported. The stepwise and continuous flow synthesis of celecoxib was achieved by means of a Claisen condensation to access 4,4,4-trifluoro-1-(4-methyl-phenyl)-butane-1,3-dione followed by a cyclo-condensation reaction with 4 sulfamidophenyl-hydrazine hydrochloride to obtain the pyrazole moiety. A batch process was developed with improved work-up and purification (90% yield). This was successfully translated to flow in yields of 90–96% with greatly shortened reaction times (20 h vs. 1 h) and reduced chemical exposure.
2020
23. A concise, rapid and high yielding flow synthesis of aryldiazonium tetrafluoroborates
Chantal Scholtz and Darren L. Riley, Arkivoc, 2020, v, 119-128
I.F. 1.003

A concise, rapid and high-yielding flow synthesis of aryl diazonium tetrafluoroborate salts is reported. The flow approach has been achieved by means of a diazotization reaction to access unstable aryl diazonium chloride salts in situ, followed by reaction with sodium tetrafluoroborate, to afford the corresponding aryldiazonium tetrafluoroborates in isolated yields of 64-100%.
* Regional issue – Organic Chemistry in South Africa*
22. Binding pose analysis of hydroxyethylamine based β-secretase inhibitors and application thereof to the design and synthesis of novel indeno[1,2-b]indole based inhibitors
C. Johan van der Westhuizen, Divan G. van Greunen, Werner Cordier, Margo Nell, Vanessa Steenkamp, André Stander, Jenny-Lee Panayides and Darren L. Riley, Arkivoc, 2020, v, 84-107
I.F. 1.003

β-Secretase (BACE1) is recognised as a target for the treatment of Alzheimer’s disease, and transition-state isosteres such as hydroxyethylamines have shown promise when incorporated into BACE1 inhibitors. A computational investigation of previously reported carbazole-based hydroxylethylamines with contradictory binding poses was undertaken using molecular dynamic simulations to rationalise the ligands preferred binding preference. Visual inspection of the confirmed binding pocket showed unoccupied space surrounding the carbazole moiety which was probed through the synthesis of seventeen ligands wherein the carbazole ring system was replaced with an indeno[1,2-b]indole ring system. The most active compound, rac-1-[benzyl(methyl)amino]-3-(indeno[1,2-b]indol-5(10H)-yl)propan-2-ol, indicated an inhibition of 91% at 10 μM against β-secretase with a cytotoxicity IC50 value of 10.51 ± 1.11 μM against the SH-SY5Y cell line.
* Regional issue – Organic Chemistry in South Africa*
21. Design and synthesis of sulphonyl acetamide analogues of quinazoline as anticancer agents
J.Khazir, B.A. Mir, M. Pandita, L. Pilcher, D. Riley and G. Chashoo, Medicinal Chemistry Research, 2020, 29, 916-925.
I.F. 1.783

A series of sulphonyl acetamide analogues were generated on the quinazoline ring through a multistep reaction starting from 2-mercapto-3H-quinazolin-4-one. The library of synthesised analogues was screened for in vitro cytotoxic activity against various human cancer cell lines such as HCT-1 and HT-15 (colon), MCF-7(Breast), PC-3 (Prostrate), SF268 (CNS) using MTT method. From the bioassay results, it was observed that even though most of the synthesised derivatives exhibited a good potency against various screened cancer cell lines, but compound 10d, 10k, and 10n were found to show very potent anticancer activity on all tested cancer cell lines with compound 10d showing IC50 value of 0.08, 0.3 and 0.55 μM on HT-29, MCF-7 and PC-3 cell lines, respectively, compound 10k showing IC50 value of 0.12, 0.03 and 0.08 μM on HCT-15, HT-29 and PC-3 cell lines, respectively, and compound 10n showing IC50 values of 0.1, 0.34, 0.52 and 0.26 on HCT-15, HT-29, MCF-7 and PC-3 cell lines, respectively.
20. Synthesis and anticancer activity of N-9– and N-7- substituted 1,2,3 triazole analogues of 2,6-di-substituted purine
J.Khazir, B.A. Mir, G.Chashoo, L. Pilcher and D. Riley, Medicinal Chemistry Research, 2020, 29, 33-45.
I.F. 1.783

A library of N-9- and N-7-substituted 1,2,3 triazole analogues were generated on the 2,6-di-substituted purine upon reaction with various substituted aromatic azides. The synthesised analogues were screened for in vitro cytotoxic activity against various human cancer cell lines like (HCT-1 (colon), THP-1 (leukaemia), IMR-32 (neuroblastoma) and A-549 (lung)). From the bioassay results, it was observed that even though most of the synthesized derivatives exhibited a good potency agains various screened cancer cell lines, but few of the analogues like 9a, 9b and 9e were found to be the most potent analogues in the series, with compound 9a showing IC50 values of 0.08 and 0.4 μM against THP-1 and A-549 cell lines, respectively.
19. Design, synthesis and anticancer evaluation of acetamide and hydrazine analogues of pyrimidine
J.Khazir, B.A. Mir, G.Chashoo, T. Maqbool, D. Riley and L. Pilcher, Journal of Heterocyclic Chemistry, 2020, 57, 1306-1318
I.F. 1.484

A library of acetamide and hydrazine analogues were generated on the pyrimidine ring through a multistep reaction starting from 5‐nitro‐pyrimidine‐4,6‐diol and pyrimidine‐4,6‐diol, respectively. The synthesized analogues were screened for in vitro cytotoxic activity against various human cancer cell lines like HCT‐1 and HT‐15 (colon), MCF‐7(breast), PC‐3 (prostrate), SF268 (CNS) using MTT method. From the bioassay results, it was observed that even though many of the synthesized derivatives exhibited a good potency against various screened cancer cell lines, compound 14a from the acetamide series was found to show potent anticancer activity on all the tested cancer cell lines with IC50 value of 0.36μM on CNS cell line and 1.6μM on HT‐21 cell line, and compound 19xxi from hydrazine series of pyrimidine showed potent activity against three tested cancer cell lines with IC50 value of 0.76μM on HT‐29 cell line, 2.6μM on HCT‐15, and 3.2μM on MCF‐7 cell line.
2019
18. A reaction energy profile and fragment attributed molecular system energy change (FAMSEC)-based protocol designed to uncover reaction mechanisms: a case study of the proline-catalysed aldol reaction
I. Cukrowski, G. Dhimba and D.L. Riley, Physical Chemistry Chemical Physics 2019, 21, 16694-16705
I.F. 3.430

A REP-FAMSEC (reaction energy profile-fragment attributed molecular system energy change) protocol designed to explain each consecutive energy change along the reaction pathway is reported. It mainly explores interactions between meaningful polyatomic fragments of a molecular system and, by quantifying energetic contributions, pin-points fragments (atoms) leading to or opposing a chemical change. Its usefulness is tested, as a case study, on the proline-catalysed aldol reaction for which a number of mechanisms have been debated for over four decades. The relative stability of S-proline conformers, their catalytic (in)activity and the superior affinity of the higher energy conformer to acetone is fully explained at atomic and molecular fragment levels, but still appealing to general chemist knowledge. We found that (i) contrary to the generally accepted view, CN-bond formation cannot be explained by the Nδ−, Cδ+ atom pair, but rather by the O-atom of acetone and its strongest inter-molecular attractive interactions with the N-atom as well as the C-atom of the COO group of proline (at this initial stage the lower energy conformer of proline is eliminated) and (ii) the following ‘first’ H-transfer from N to O atoms of the proline moiety is nearly energy-free even though initially the H-atom interacts three times stronger with the N- than O-atom; a full explanation of this phenomenon is provided.
17. Novel N-benzylpiperidine carboxamide derivatives as potential cholinesterase inhibitors for the treatment of Alzheimer’s disease
D.G. van Greunen, C.J. van der Westhuizen, W. Cordier, M. Nell, A. Stander, V. Steenkamp, J.-L. Panayides, D.L. Riley. European Journal of Medicinal Chemistry 2019, 179, 680-693.
I.F. 5.573

A series of fifteen acetylcholinesterase inhibitors were designed and synthesised based upon the previously identified lead compound 5,6-dimethoxy-1-oxo-2,3-dihydro-1H-inden-2-yl 1-benzylpiperidine-4-carboxylate (5) which showed good inhibitory activity (IC50 0.03 ± 0.07 μM) against acetylcholinesterase. A series of compounds were prepared wherein the ester linker in the original lead compound was exchanged for a more metabolically stable amide linker and the indanone moiety was exchanged for a range of aryl and aromatic heterocycles. The two most active analogues 1-benzyl-N-(5,6-dimethoxy-8H-indeno[1,2-d]thiazol-2-yl)piperidine-4-carboxamide (28) and 1-benzyl-N-(1-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl) piperidine-4-carboxamide (20) afforded in vitro IC50 values of 0.41 ± 1.25 and 5.94 ± 1.08 μM, respectively. In silico screening predicts that 20 will be a blood brain-barrier permeant, and molecular dynamic simulations are indicative of a close correlation between the binding of 20 and the Food and Drug Administration-approved cholinesterase inhibitor donepezil (1).
16. Landscape and opportunities for active pharmaceutical ingredient manufacturing in developing African economies
D.L. Riley, I. Strydom, R. Chikwamba and J.-L. Panayides, Reaction Chemistry and Engineering 2019, 4, 457-489
I.F. 3.440

Africa is one of the world’s fastest growing economies, with South Africa having the fifth highest worldwide pharmaceutical expenditure per capita. In recent years, several companies have considered regional pharmaceutical production but have failed to make the investment, in stark contrast to the massive growth in pharmaceutical production in other BRICS countries. Major constraints identified have been the small local market, lack of skills, and an export-averse culture, which have prevented regional manufacturers from achieving the economies of scale that are essential to survive in a global market. In contrast, the pharmaceutical industry is undergoing a revolutionary change in manufacturing, with the potential to switch from batch manufacturing to continuous flow processing. The possibility of applying this new pharmaceutical business model in emerging markets will open the door for dramatic changes in regional commercial manufacturing. Advances in cloud computing, automation and system unification are paving the way for continuous active pharmaceutical ingredient production with integrated digital connectivity. This review will highlight the opportunities that exist in the localization of cutting-edge manufacturing technologies; in order to show the potential application of fundamental process research key production examples relevant to the region will be provided.
*2019 Emerging Investigators issue*
15. Immobilized tetrakis(triphenylphosphine)palladium(0) for Suzuki–Miyaura coupling reactions under flow conditions
G.V. Ramaotsoa, I. Strydom, J.-L. Panayides, D.L. Riley Reaction Chemistry and Engineering 2019, 4, 372-382
I.F. 3.440

An immobilized triphenylphosphine scaffold was prepared by precipitation polymerization and functionalized to afford a cost-effective source of solid-supported tetrakis(triphenylphosphine)-palladium(0). The catalyst was characterised and used to perform biphasic Suzuki–Miyaura cross-coupling reactions using a packed-bed reactor under flow conditions. The approach afforded comparable yields to those obtained under batch conditions with a single pass through the packed-bed reactor (1 h vs. 18 h). The use of a recycling system was investigated on a model reaction and found to afford close to quantitative conversion within 3 hours.
*2019 Emerging Investigators issue*
2018
14. Mild and selective reductions of aldehydes utilising sodium dithionite under flow conditions.
N.C. Neyt, D.L. Riley Beilstein Journal of Organic Chemistry 2018, 14, 1529-1536
I.F. 2.595

We recently reported a novel hybrid batch–flow synthesis of the antipsychotic drug clozapine in which the reduction of a nitroaryl group is described under flow conditions using sodium dithionite. We now report the expansion of this method to include the reduction of aldehydes. The method developed affords yields which are comparable to those under batch conditions, has a reduced reaction time and improved space-time productivity. Furthermore, the approach allows the selective reduction of aldehydes in the presence of ketones and has been demonstrated as a continuous process.
13. Approaches for performing reductions under continous flow conditions.
D. L. Riley, N.C. Neyt. Synthesis, 2018, 50 (14), 2707-2720
I.F. 2.867

A concise overview of approaches to perform reductions of various functionalities including aldehydes, ketones, esters, imines, nitriles, nitro groups, alkenes and alkynes under continuous-flow conditions are highlighted and discussed in this short review.
12. Batch-flow hybrid synthesis of the anti-psychotic clozapine.
N.C. Neyt, D. L. Riley. Reaction Chemistry and Engineering, 2018, 3, 17-24.
I.F. 4.010


The development of batch–flow hybrid processes is becoming an attractive prospect through which chemists can make use of the best aspects of both technologies. We have reported the implementation of an on-the-fly purification by trituration which can also be utilised to perform solvent swaps. We have demonstrated this concept through the synthesis of the antipsychotic clozapine. In addition, we report a novel means of performing a reduction of an aryl nitro group under flow conditions and an overall improved process route for the total synthesis of clozapine.
2017
11. Targeting Alzheimer’s disease by investigating previously unexplored chemical space surrounding the cholinesterase inhibitor donepezil.
D.G. van Greunen, W. Cordier, M. Nell, C. van der Westhuyzen, V. Steenkamp, J. Panayides, D.L. Riley. European Journal of Medicinal Chemistry 2017, 127, 671-690.
I.F. 4.816

A series of twenty seven acetylcholinesterase inhibitors, as potential agents for the treatment of Alzheimer’s disease, were designed and synthesised based upon previously unexplored chemical space surrounding the molecular skeleton of the drug donepezil, which is currently used for the management of mild to severe Alzheimer’s disease. Two series of analogues were prepared, the first looking at the replacement of the piperidine ring in donepezil with different sized saturated N-containing ring systems and the second looking at the introduction of different linkers between the indanone and piperidine rings in donepezil. The most active analogue 5,6-dimethoxy-1-oxo-2,3-dihydro-1H-inden-2-yl 1-benzylpiperidine-4-carboxylate (67) afforded an in vitro IC50value of 0.03 ± 0.07 μM against acetylcholinesterase with no cytotoxicity observed (IC50of >100 μM, SH-SY5Y cell line). In comparison donepezil had an IC50 of 0.05 ± 0.06 μM and an observed cytotoxicity IC50 of 15.54 ± 1.12 μM. Molecular modelling showed a strong correlation between activity and in silico binding in the active site of acetylcholinesterase.
2016
10. New syntheses of (±)-tashiromine and (±)-epitashiromine via enaminone intermediates
Riley, D. L.; Michael, J. P.; de Koning, C. B. Beilstein Journal of Organic Chemistry 2016, 12, 2609–2613.
I.F. 2.337

A synthesis of the naturally occurring indolizidine alkaloid (±)-tashiromine and its unnatural epimer (±)-epitashiromine are demonstrated through the use of enaminone chemistry. The impact of various electron-withdrawing substituents at the C-8 position of the indolizidine core on the preparation of the bicyclic system is described.
9. Design and synthesis of ring C opened analogues of α-santonin as potential anticancer agents
J. Khazir, B.A. Mir, L.A. Pilcher, D.L. Riley, G. Chashoo, A. Islam, A.K. Saxena, H.M.S. Kumar, Medicinal Chemistry Research, 2016, 25: 2030-2041.
I.F. 1.277

Here we describe ring opening reaction of a novel halo triene derivative viz., (3S, 5aS)-8-chloro-3a, 4, 5, 5a-tetrahydro-3, 5a, 9-trimethylnaphtho [1, 2-b] furan-2(3H)-one of α-santonin upon nucleophillic attack with alcohols. Halo-triene was synthesized from α-santonin upon reaction with vilsmeier reagent. The synthesized compounds from ring opening reaction were evaluated for anticancer activity against a panel of four human cancer cell lines (A-549, THP-1, HCT-15, and IMR-13). Most of the compounds exhibited promising anticancer activity against all cancer cells in vitro; however compound. 3d with benzyl substitution showed most potent anticancer activity with an IC50 value of 0.3, 0.51, 0.6 and 0.23 μM against A-549, THP-1, HCT-116 and IMR-13 cell lines respectively.
8. An improved process for the preparation of tenofovir diisoproxil fumarate
D.L. Riley, D.R. Walwyn, C.D. Edlin, Organic Process Research and Development, 2016, 20, 742-750
I.F. 2.857

The current three-step manufacturing route for the preparation of tenofovir disoproxil fumarate (1) was assessed and optimized leading to a higher yielding, simpler, and greener process. Key improvements in the process route include the refinement of the second stage through the replacement of the problematic magnesium tert-butoxide (MTB) with a 1:1 ratio of a Grignard reagent and tert-butanol. The development of a virtually solvent-free approach and the establishment of a workup and purification protocol which allows the isolation of a pure diethyl phosphonate ester (8) was achieved.
2015
7. Design, synthesis and anticancer activity of Micheal-type thiol adducts of α-santonin analogue with exocyclic methylene
J. Khazir, D.L. Riley, G. Chashoo, B.A. Mir, D. Liles, A. Islam, S.K. Singh, R.A. Vishwakarma, L.A. Pilcher, European Journal of Medicinal Chemistry, 2015, 101, 769-779.
I.F. 3.902

A series of Michael-type analogues were generated on the C-ring of α-santonin (α-methylene-γ-butyrolactone) upon reaction with various thiols. All the thiol adducts synthesized were evaluated for their anticancer activity against four human cancer cell lines (PC-3, HCT-15, A-549 and MCF-7). Bioassay results indicated that even though most of the synthesized compounds exhibited a good anticancer activity against various cancer cells in vitro, some of the compounds like 9e, 9g and 9q were found to be the most promising analogues in this series, with compound 9e showing IC50 values of 1.5 μM, 0.6 μM, 2.4 μM and 1.2 μM on PC-3, MCF-7, A-549 and HCT-116 cell lines respectively. Further, flow cytometry studies showed that MCF-7 cells treated with the compounds 9e, 9g and 9q were arrested in the sub G1 phase of the cell cycle in a concentration dependent manner. These lead molecules were further studied for NF-κB, p65 transcription factor inhibitory activity which confirmed concentration dependent inhibition against NF-κB, p65 with analogue 9e showing 57% inhibition at 2 μM, 9g showing 62% inhibition at 3 μM and 9q showing 54% inhibition at 2 μM concentration.
6. An inquiry-based practical curriculum for organic chemistry as preparation for industry and postgraduate research
L.A. Pilcher, D.L. Riley, K.C. Mathabathe, M. Potgieter, South African Journal of Chemistry, 2015 68, 236-244
I.F. 0.667

This paper describes the development of a new practical curriculum for third-year organic chemistry to replace the recipe-based approach typically used in undergraduate teaching laboratories. The new curriculum consists of an inquiry-based project set in a simulated industrial context preceded by two scaffolding experiments to prepare students for the task. The industrial project requires students to evaluate experimentally three multi-step synthetic routes to a given target based on cost, technical challenge and environmental impact in order to make a recommendation as to which route the ‘company’ should use to synthesize the compound. The project equips students with technical skills suitable for both postgraduate research and industry, and develops metacognition and understanding through the use of the jig-saw cooperative learning strategy and reflection. The students were found to engage with the practical work at a deep intellectual level, demonstrating that contextualized inquiry-based laboratory teaching afforded an improved quality of learning. In addition, the reported practical curriculum made a difficult subject accessible and even popular, to some measure grew the students’ ability in all desired graduate attributes and resulted in the establishment of a professional identity for individual students.
2014
5. Role of plants in anticancer drug discovery
J. Khazir, B.A. Mir, L. Pilcher, D.L. Riley, Phytochemistry Letters, 2014, 7, 173-181
I.F. 1.450

Cancer is one of the major causes of death and the number of new cases, as well as the number of individuals living with cancer, is expanding continuously. Worldwide the alarming rise in mortality rate due to cancer has fuelled the pursuit for effective anticancer agents to combat this disease. Finding novel and efficient compounds of natural origin has been a major point of concern for research in the pharmaceutical sciences. Plants have been seen to possess the potential to be excellent lead structures and to serve as a basis of promising therapeutic agents for cancer treatment. Many successful anti-cancer drugs currently in use or their analogues are plant derived and many more are under clinical trials. This review aims to highlight the invaluable role that plants have played, and continue to play, in the discovery of anticancer agents.
*Phytochemistry Letters Top Cited Author 2014*
4. Anticancer agents from diverse natural sources
J. Khazir, D.L. Riley, L.A. Pilcher, P. De-Maayer and B.A. Mir Natural Product Communications, 2014, 9, 1655-1669.
I.F. 0.906
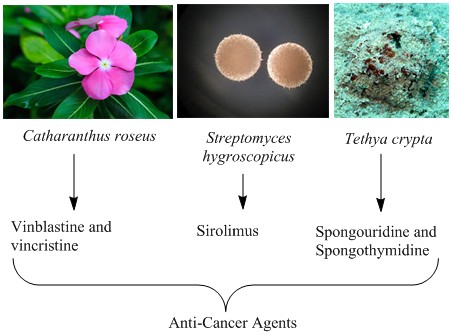
This review attempts to portray the discovery and development of anticancer agents/drugs from diverse natural sources. Natural molecules from these natural sources including plants, microbes and marine organisms have been the basis of treatment of human diseases since the ancient times. Compounds derived from nature have been important sources of new drugs and also serve as templates for synthetic modification. Many successful anti-cancer drugs currently in use are naturally derived or their analogues and many more are under clinical trials. This review aims to highlight the invaluable role that natural products have played, and continue to play, in the discovery of anticancer agents.
3. A process for the preparation of (R)-9-[2-(phosphonomethoxy)propyl]adenine (PMPA)
D.L. Riley, D.R. Walwyn, C.D. Edlin, WO2014033688 A1

This invention relates to a process for the preparation of (R)-9-[2- (phosphonometh-oxy)propyl]adenine (PMPA). This invention also relates to the preparation of Tenofovir disoproxil fumarate (TDF), a compound which can be produced from PMPA. The claimed process comprises reacting compound (6) with compound (7) in presence of 2,2,6,6- tetramethylpiperidinylmagnesium to compound (2), which is hydrolysed to (3).
2011
2. Oxepines and Azepines
D.L. Riley and W.A.L. van Otterlo, in “Heterocycles in Natural Product Synthesis”, Editors: K.C. Majumdar and S.K. Chattopadhyay, Wiley-VCH Verlag, Weinheim Germany, 2011, 537-568.

Filling a gap on the market, this handbook and ready reference is unique in its discussion of the usefulness of various heterocyclic systems in the synthesis of natural products. Clearly structured for easy access to the information, each chapter is devoted to a certain class of heterocycle, providing a tabular presentation of the natural products to be covered containing the particular heterocyclic ring system along with their biological profile, occurrence and most important physical properties, backed by the appropriate references. In addition, the application of the heterocyclic system to the synthesis of natural products are covered in detail. Of great interest to organic, natural products, medicinal and biochemists, as well as those working in the pharmaceutical and agrochemical industry.
2009
1. Formal synthesis of (5R,8R8aS)-indolizidine 209I via enaminones incorporating Weinreb amides
C.B. de Koning, J.P. Michael, D.L. Riley, Heterocycles, 2009, 79, 935-953
I.F. 1.165

A formal enantioselective synthesis of the amphibian alkaloid (5R,8R,8aS)-(–)-indolizidine 209I (6) is reported. Control of the absolute stereochemistry at C-5 resulted from application of the Davies procedure, which entails stereoselective conjugate addition of (R)-(+)-N-benzyl-1-phenylethylamine to tert-butyl (E)-hex-2-enoate. The resulting chiral adduct 26 was converted in eight steps into a pivotal enaminone incorporating a Weinreb amide, the inherent nucleophilicity of which was exploited in a cyclisation that yielded the key bicyclic intermediate (5R)-N-methoxy- N-methyl-5-propyl-1,2,3,5,6,7-hexahydroindolizine- 8-carboxamide (38). Stereoselective catalytic hydrogenation of the alkene bond, reaction of the Weinreb amide with ethylmagnesium bromide, and epimerisation of the resulting ketone completed the formal synthesis of the target alkaloid.